闵行区CDE eCTD名称
关键词: 闵行区CDE eCTD名称 eCTD

 2025.12.20
2025.12.20
文章来源:
技术壁垒与兴市场挑战非洲和东南亚guo家逐步采纳eCTD,但其IT基础设施薄弱导致实施进度滞后。欧盟通过“eCTD全球化倡议”提供技术援助,帮助兴市场建立验证体系和培训中心。跨国yao企需针对不同区域定制递交策略,例如在模块1附加本地稳定性数据。监管科学与创激励eCTD支持真实世界证据(RWE)和适应性临床试验设计的整合,加速创yao上市。EMA的PRIME计划为突破性疗法提供eCTD快su通道,允许分阶段提交模块数据。孤儿yao和儿科yao的eCTD序列可享受费用减免和优先审评。供应链安全与审计追踪eCTD的XML主干文件记录所有提交版本,支持供应链问题的追溯分析。原料yaoCEP持有者需及时更变更信息,确保下游制剂厂商获取数据。区块链技术试点用于追踪eCTD数据流,防止篡改和未授权访问。文化差异与实施障碍部分南欧guo家偏好传统纸质流程,导致eCTD推广阻力较大。EMA通过多语种培训材料和区域协调员制度促进文化适应。行业需调整管理思维,将eCTD从“合规负担”转化为“竞争优势”。 加拿大IND注册申报相关技术支持。闵行区CDE eCTD名称
2020年暴发后,FDA进一步推动电子化进程,例如允许远程电子签章和临时放宽部分格式要求,但验证标准(如PDF版本、书签链接有效性)并未降低。这一时期的实践为eCTD在紧急审批中的灵活性提供了案例,也凸显了其作为危机应对工具的价值。 尽管美国尚未部署eCTD V4.0,但其技术方向已明确:支持医疗器械和保健品申报、增强数据可复用性、优化审评系统与人工智能的集成。此外,区块链技术在电子签章和数据溯源中的应用探索,可能成为下一阶段升级的重点。太仓INDeCTD推荐eCTD验证标准相关技术支持。
从纸质到电子的历史过渡 2017年前,美国允许纸质与eCTD并行提交,但此后逐步淘汰纸质通道,保留紧急情况下的例外审批。2020年电子化后,所有IND、NDA、ANDA和DMF强制采用eCTD格式。 系统平台升级 FDA通过“yao品业务应用系统”和“yao品eCTD注册系统”实现电子资料接收、受理与审评的全流程数字化。2022年系统增自动推送受理文书和短信提醒功能,减少人工干预。 电子文档结构优化 美国eCTD采用分层文件夹结构,例如化学yao品的模块1-5分别对应行政文件、总结报告、质量数据等。2020年后增“临床试验数据库”装盒要求,强化数据可追溯性。
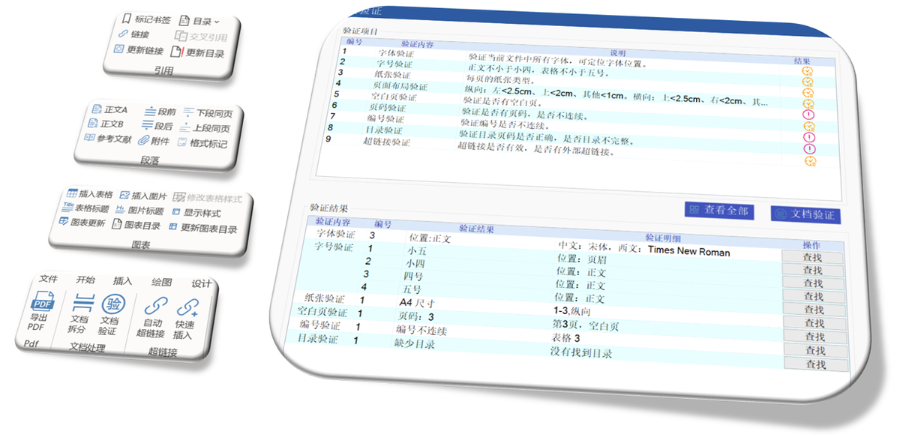
赋悦Word插件自主研发Word插件kuai速编辑:整合word常用功能按钮,避免频繁切换菜单;内置标题、段落、文字、目录、超链接等的格式和样式,可kuai速设置和更文档的格式kuai速链接:双击或者拖拽的方式,制作文本超链接或者题注超链接;可搜索全文关键字,自动制作超链接文档拆分:可根据不同的条件将word文件颗粒化,如分节符、页眉、页脚、页码范围和自定义页码等PDF转换:WORD转PDF,自动判断是否生成书签,自动镶嵌所有字体,生成PDFkuai速网页浏览的PDF,确保生成的PDF所有属性符合法规要求文档验证:验证文档的字体、字号、纸张、页面布局、空白页、页码、编号、目录、超链接等,并且可以ding位验证结果可定制:可根据用户需求定制格式和样式模板。 美国ANDA注册申报相关技术支持。
欧盟eCTD的递交途径与技术要求不同审评程序对应不同递交渠道:集中程序(CP)通过EMA的eSubmissionGateway或WebClient提交,分散程序(DCP)和互认程序(MRP)则需使用欧盟通用提交门户(CESP)。文件结构需严格遵循模块化要求,例如CEP申请需包含模块1(行政文件)、模块2(质量概述)和模块3(技术文档),且XML主干文件须符合EDQM的特定命名规则。此外,所有PDF文件需无密码保护、可全文检索,并嵌入层级书签以支持快su审阅。CEP申请的eCTD递交特殊性CEP程序自2018年起强zhi采用eCTD格式,重点评估原料yao是否符合欧洲yao典标准。其模块1需包含EDQM申请表、简历及变更说明表,模块2需使用EDQM提供的质量概述模板,模块3则按CTD格式组zhi。CEP与ASMF(活性物质主文件)的主要区别在于性:CEP无需关联上市许可,且审评由EDQM完成。澳大利亚eCTD验证标准相关技术支持。江苏中国eCTD文件如何制作
美国API的DMF申报相关技术支持。闵行区CDE eCTD名称
美国药物主文件(Drug Master File, DMF)是向FDA提交的机密技术文件,用于支持药品生产、质量控制及合规性审查。以下为申报的要点和流程总结: DMF概述与类型 定义与作用 DMF是药品生产全过程的详细档案,包含原料药、辅料、包装材料等的生产设施、工艺、质量控制等信息,供制剂厂商引用以支持其注册申请。其意义在于保护企业机密的同时,满足FDA对供应链透明度的要求。 DMF类型 Ⅱ类:原料药、中间体及制剂(如微生物外泌体、细胞株等生物制品均属此类)。 Ⅲ类:包装材料。 Ⅳ类:辅料、着色剂等添加剂。 Ⅴ类:非临床/临床数据等特殊信息(需FDA预先批准)。 注:Ⅰ型(生产设施与人员)已于2000年停用。闵行区CDE eCTD名称

- 高新区国内注册eCTD格式 2025-12-19
- 静安区仿制药eCTD哪家好 2025-12-19
- 苏州CDE eCTD服务放心可靠 2025-12-18
- 芜湖eCTD性价比 2025-12-18
- 高新区INDeCTD使用 2025-12-18
- 工业园区中国eCTD服务价格 2025-12-18
- 上海国内注册eCTD服务价格 2025-12-18
- 芜湖化学药品eCTD发布软件 2025-12-18




- 01 立卓无线扫描枪多少钱
- 02 中国台湾证卡打印机制造厂家
- 03 凤阳智能化管控弱电智能化集成供应
- 04 安顺智能一站式闭环营销常见问题
- 05 湖北工业4.0工控机价格
- 06 上海工业级物联网服务商
- 07 视觉方案厂家
- 08 智能**智能撰写平台
- 09 江苏工业级彩膜面板UV打印机厂商
- 10 辽宁电动合唱话筒升降器上门安装